


























































































































































Introduction to Martini
- Details
- Last Updated: Friday, 27 January 2023 10:16
The Martini force field is a coarse-grain (CG) force field suited for molecular dynamics simulations of biomolecular systems. The force field has been parametrized in a systematic way, combining top-down and bottum-up strategies: Non-bonded interactions are based on the reproduction of experimental partitioning free energies between polar and apolar phases of a large number of chemical compounds, whereas bonded interactions are derived from reference all-atom simulations.
The model uses a four-to-one mapping, i.e. on average four heavy atoms and associated hydrogens are represented by a single interaction center. In order to keep the model simple, only four main types of interaction sites are defined: polar, non-polar, apolar, and charged. Each particle type has a number of subtypes, which allow for an accurate representation of the chemical nature of the underlying atomistic structure.
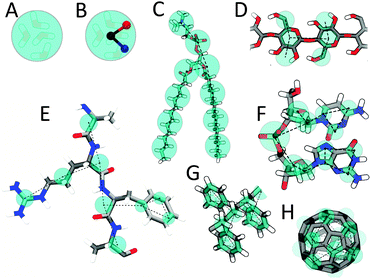
Martini mapping examples of selected molecules: (A) Standard water particle representing four water molecules, (B) Polarizable water molecule with embedded charges, (C) DMPC lipid, (D) Polysaccharide fragment, (E) Peptide, (F) DNA fragment, (G) Polystyrene fragment, (H) Fullerene molecule. In all cases Martini CG beads are shown as cyan transparent beads overlaying the atomistic structure.
Currently topologies are available for many lipids and surfactant molecules, including cholesterol, and for all amino acids and nucleotides as well as for a variety of sugars, polymers, and nanoparticles. Scripts are furthermore available to build topologies for arbitrary peptides, proteins, polynucleotides, to add elastic networks, and to move between coarse-grained and atomistic representations.
Key Martini papers include:
-
C. Hilpert, L. Beranger, P.C.T. Souza, P.A. Vainikka, V. Nieto, S.J. Marrink, L. Monticelli, G. Launay. Facilitating CG Simulations with MAD: The MArtini Database Server. J. Chem. Inf. Model., 2023, online. doi:10.1021/acs.jcim.2c01375
-
S.J. Marrink, L. Monticelli, M.N. Melo, R. Alessandri, D.P. Tieleman, P.C.T. Souza. Two decades of Martini: Better beads, broader scope. Wires Computational Molecular Science, 13(1), e1620, 2023. https://wires.onlinelibrary.wiley.com/doi/full/10.1002/wcms.1620
-
R. Alessandri, J. Barnoud, A.S. Gertsen, I. Patmanidis, A.H. de Vries, P.C.T. Souza, S.J. Marrink. Martini 3 Coarse-Grained Force Field: Small Molecules. Advanced Theory and Simulations 5, 2100391, 2022. https://onlinelibrary.wiley.com/doi/full/10.1002/adts.202100391
-
R. Alessandri, F. Grünewald, S.J. Marrink. The Martini Model in Materials Science, Adv. Materials 33:2008635, 2021. https://doi.org/10.1002/adma.202008635
-
P.C.T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, ... S.J. Marrink. Martini 3: a general purpose force field for coarse-grained molecular dynamics. Nature Methods 18:382–388, 2021. doi:org/10.1038/s41592-021-01098-3
-
B.M.H. Bruininks, P.C.T. Souza, S.J. Marrink. A Practical View of the Martini Force Field. Biomolecular Simulations, 105-127, 2019. doi:10.1007/978-1-4939-9608-7_5 , pdf-reprint.
-
S.J. Marrink, D.P. Tieleman. Perspective on the Martini model. Chem. Soc. Rev., 42:6801-6822, 2013. open access
-
D.H. de Jong, G. Singh, W.F.D. Bennett, C. Arnarez, T.A. Wassenaar, L.V. Schäfer, X. Periole, D.P. Tieleman, S.J. Marrink. Improved parameters for the Martini coarse-grained protein force field, JCTC, 9:687–697, 2013. open access
-
L. Monticelli, S.K. Kandasamy, X. Periole, R.G. Larson, D.P. Tieleman, S.J. Marrink.The MARTINI coarse grained forcefield: extension to proteins. JCTC, 4:819-834, 2008. abstract.
-
S.J. Marrink, H.J. Risselada, S. Yefimov, D.P. Tieleman, A.H. de Vries. The MARTINI forcefield: coarse grained model for biomolecular simulations. JPC-B, 111:7812-7824, 2007. abstract
More details about the Martini model can be found in the publications listed on Google Scholar's MARTINI page