Molecular Modeling Practical
This tutorial provides a short introduction to the set-up and running of Molecular Dynamics (MD) simulations of small peptides. The protocol used is a suitable starting point for investigation of peptides, provided that the system does not contain non-standard groups. At the end of the tutorial, the student should know the steps involved in setting up and running a simulation, including some reflection on the choices made at different stages. Besides, the student should know how to perform quality assurance checks on the simulation results and have a feel for methods of analysis to retrieve information.
Including NMR Restraints to the Molecular Dynamics Model
Molecular dynamics (MD) simulations consist of three stages: First, the input data (the simulation system) have to be prepared. Second, the production simulation can be run and finally the results have to be analysed and be put in context. Although the second stage is the most computationally demanding step, with simulations commonly running for several months, the most laborious steps are the preparation and the analysis. This part of the tutorial shows how restraints from Nuclear Magnetic Resonance (NMR) experiments can be incorporated in a model, and prompts comparison of the behavior with and without the restraints. Two simulations will be set up: one starting from a conformation close to the PDB structure, and one from an unfolded structure.
This tutorial uses Gromacs (http://www.gromacs.org) version 2016.1 and most of the commands given are specific to this package, although the structure is much the same for earlier (in particular versions 5) or later versions. This part of the tutorial assumes that you are familiar with setting up, running, and analyzing MD simulations as done in the earlier parts of this tutorial. You may need to refer to those earlier sections, and also see the workflow(s).
{kind=link}
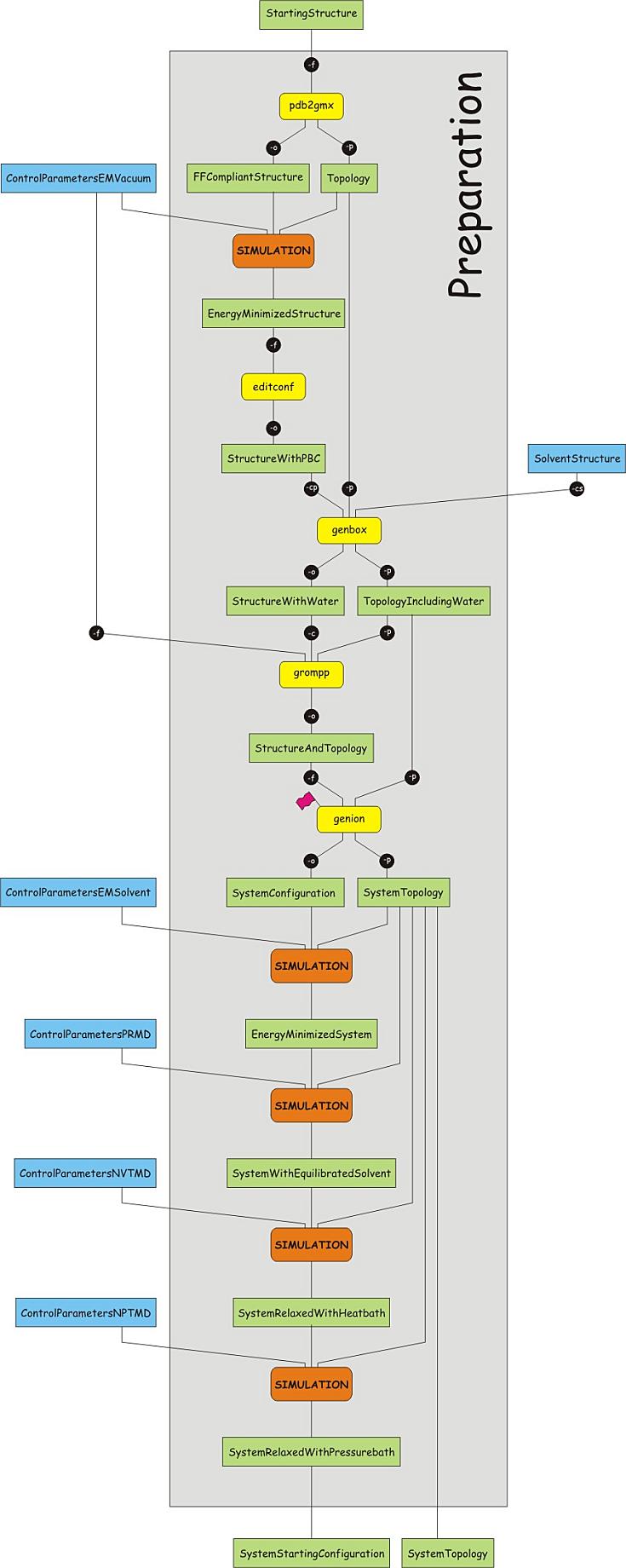
Reminder of general structure of GROMACS programs and position of the restraints
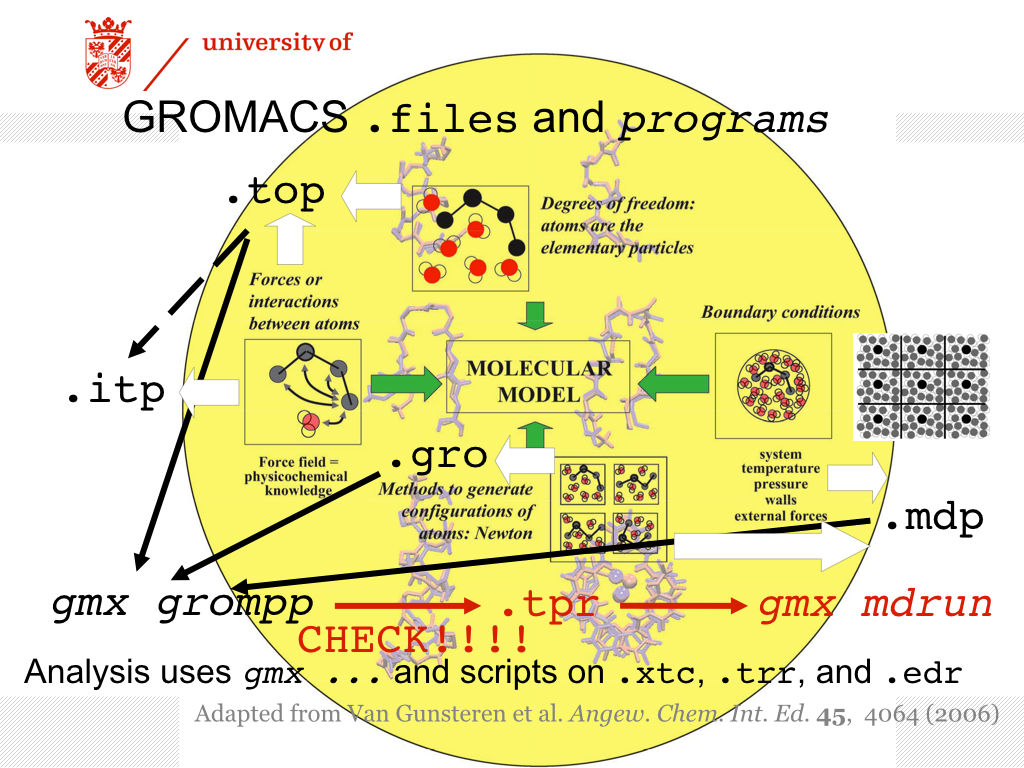
The Figure above serves as an overview of the files required for using the GROMACS suite of programs for an MD simulation. The definition of the molecule and the system (how many molecules of what type are present) reside in a file with an extension .top. To enhance readability, the .top file may refer to other files containing information, with extension .itp, usually these contain the specification of the interactions (i.e. the force field). These files are all readable, you may open them with an editor, e.g. gedit.
In this part of the tutorial, the .itp file of the peptide will be augmented, essentially specifying additional interactions between selected atoms. The interactions are based on experimental observations from NMR. The reasons for and purposes of using restraints may be various, and one may argue about the meaning of the simulations with the restraints. Here, we will first show how restraints can be incorporated, and what the consequences are. You are then encouraged to think about the meaning of applying restraints.
Preliminaries
The starting point for this part of the tutorial is a completed set-up of the Trp-cage peptide, at the point where the system is ready for a production simulation, see the entry point for the production run.
We will need to make sure our earlier results are not affected by our new simulation. We will therefore start by making a new directory and copying all files that we need for the new simulation. Make sure your prompt is in the folder/directory that contains your data for the production simulation, and type:
mkdir WITHBBRESTRAINTS
cp protein.top WITHBBRESTRAINTS/.
cp protein-NPT.gro WITHBBRESTRAINTS/.
cd WITHBBRESTRAINTS
Incorporating distance restraints in the simulation
 Computer simulations give exquisite control over
all aspects of the model. Here, we add extra interactions between
atoms, i.e. interactions that are not part of the force field
and not part of the temperature and pressure regulation.
In this case, the extra interactions are based on NMR data that
indicate that some atoms are on average close to each other in
space. The intensity of so-called NOE (Nuclear Overhauser Effect)
peaks in selected NMR spectra provides a measure for the distance
that pairs of (equivalent) atoms have with respect to each other.
Unfortunately, the exact distance cannot be read off these data,
but a reasonable range of the distance can be given.
This leads to the idea of distance restraints: the distance between
two atoms should be within certain limits. In MD simulations, the
limits are not strictly imposed. That would correspond to a very
large force when two atoms move outside the given range, and likely
result in numerical instability of the MD algorithm. Instead,
the potential energy smoothly increases when the distance between
atoms moves outside the prescribed range. Usually, the potentials used
to impose restraints are simple harmonic potentials, and GROMACS
implements these. The user needs to specify which pairs of atoms are
involved in the distance restraint and what the range of reasonable
distances is. This list is added to the topology file (.top or .itp).
In addition, the user must specify how strongly the restraint is enforced,
i.e., how quickly the energy rises when the distance between the atoms
is outside the specified range. This is in part written in the list of
distance restraints in the topology file, and in part governed by the
settings in the md control file (.mdp).
Computer simulations give exquisite control over
all aspects of the model. Here, we add extra interactions between
atoms, i.e. interactions that are not part of the force field
and not part of the temperature and pressure regulation.
In this case, the extra interactions are based on NMR data that
indicate that some atoms are on average close to each other in
space. The intensity of so-called NOE (Nuclear Overhauser Effect)
peaks in selected NMR spectra provides a measure for the distance
that pairs of (equivalent) atoms have with respect to each other.
Unfortunately, the exact distance cannot be read off these data,
but a reasonable range of the distance can be given.
This leads to the idea of distance restraints: the distance between
two atoms should be within certain limits. In MD simulations, the
limits are not strictly imposed. That would correspond to a very
large force when two atoms move outside the given range, and likely
result in numerical instability of the MD algorithm. Instead,
the potential energy smoothly increases when the distance between
atoms moves outside the prescribed range. Usually, the potentials used
to impose restraints are simple harmonic potentials, and GROMACS
implements these. The user needs to specify which pairs of atoms are
involved in the distance restraint and what the range of reasonable
distances is. This list is added to the topology file (.top or .itp).
In addition, the user must specify how strongly the restraint is enforced,
i.e., how quickly the energy rises when the distance between the atoms
is outside the specified range. This is in part written in the list of
distance restraints in the topology file, and in part governed by the
settings in the md control file (.mdp).
The NMR data lead to 169 distance restraints, but we will use only some of the restraints associated with backbone H-atoms. These have been collected in a file, called disre-bb.itp. Download this file to the working directory WITHBBRESTRAINTS. We need to make sure the distance restraint file is read by the GROMACS preprocessor. To this end, the topology file protein.top must be edited. Open the file protein.top in an editor (e.g. gedit):
gedit protein.top
and add a line to it, reading:
#include "disre-bb.itp"
This line must be near the end of the file, after all bonded interactions have been defined, but before the directive [system]. The best place is probably straight after the list of the bonded interactions. Also, download the control parameter file md-res.mdp to the working directory WITHBBRESTRAINTS. Now, prepare the .tpr file for a 2 ns run with the restraints, starting from the same structure as the previous production run and set the simulation going:
gmx grompp -v -f md-res.mdp -c protein-NPT.gro -p protein.top
gmx mdrun >& md.job &
NOTE: While the simulation is running, study the next section that describes how the distance restraints are given in the Protein Data Bank and how they are communicated to the GROMACS program.
When the simulation has finished, you can run all the analysis that was done for the production run and compare the behavior of the simulations with and without the distance restraints. Simply follow the instructions of the analysis section and compare the outputs of the simulation with distance restraints to the results obtained from the production run.
However, in view of the available time, you may first want to complete the assignment, which asks you to investigate whether distance restraints can help the Trp-cage to fold in a relatively short time from an unfolded structure. In that case, proceed to the assignment after you have studied the next section about the way the distance restraints can be added to an MD simulation.
Specification of NMR Restraints as deposited in the Protein Data Bank
 The Protein Data Bank is a repository for structures obtained by
X-ray and NMR spectroscopy. Apart from final coordinates, other
data may also be deposited, such as distance restraints.
The deposited data adhere to standards that are often used in
different softwares for data analysis and/or modeling and calculations
using the deposited data.
The Protein Data Bank is a repository for structures obtained by
X-ray and NMR spectroscopy. Apart from final coordinates, other
data may also be deposited, such as distance restraints.
The deposited data adhere to standards that are often used in
different softwares for data analysis and/or modeling and calculations
using the deposited data.
One of the formats used for NMR distance restraints, and the format most easily converted to distance restraints in the GROMACS molecular dynamics simulation package is the .mr format. This format for describing NOE distance restraints is described at the website of the CNS suite of programs. CNS stands for Crystallography and NMR System.
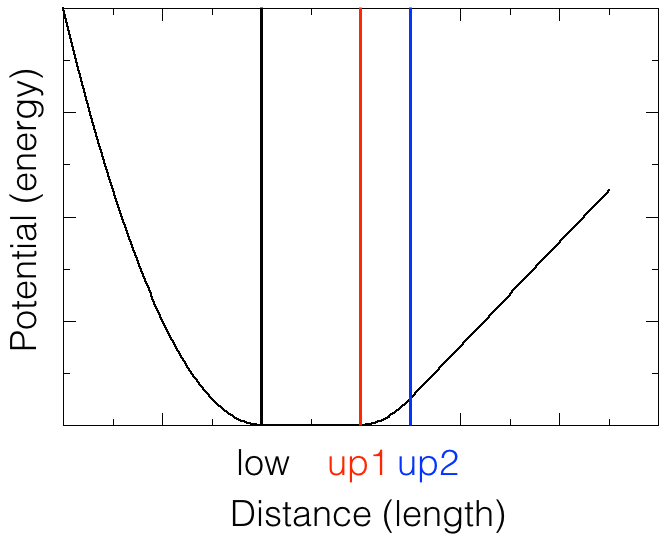
The file disre-bb.itp contains entries for each distance restraint added in the simulation. Here, we choose to implement the distance restraints as special types of bonds, that use the so-called flat-bottom potential. A sketch of this potential is shown in the figure below. In the region between low and up1, the potential is flat: the atom pair does not feel any extra force. In the region below low and between up1 and up2, the potential is a harmonic function, which means that the force is proportional to the deviation of the distance from low or up1; the force is such that the distance wants to change in the direction of low or up1. The strength of the force depends on the deviation of the distance and on the force constant. The force constant may be set according to the strength of the NOE. If the distance is larger than up2, the potential increases linearly, and the force is constant, pulling the atoms closer together.
The structure of the file disre-bb.itp is the following: the first line is the directive for bonds (compare to other directives, for example for bonds, angles, dihedrals in the topology (protein.top)); the second line is a comment line helping you to remember the order and meaning of the parameters used in the directive; the third line holds the specification of the special bond used to implement one of the distance restraints. The first entry in the file disre-bb.itp matches one of the entries in the file CNS file 1l2y.mr downloaded from the PDB and is repeated below.
[ bonds ]
; atom i j func low up1 up2 fc
18 37 10 0.0 0.30 0.35 120.0
Identify which entry in the file 1l2y.mr is matched by the first entry in the file disre-bb.itp. Why does it make sense to set the LOWER limit to 0.0, rather than the value given in the CNS file?