Protein-Ligand Binding
- Details
-
Last Updated: Wednesday, 25 January 2023 14:23
Binding of ligands to proteins is a challenging area for coarse-grained models. Most binding pockets require specific hydrogen bonding patterns and a neat fit of the ligand. With a CG model such as Martini that lacks directional hydrogen bonds and can not represent the fine details of the packing, ligand binding may require multi-scale methods.
However, less specific binding may be modeled with Martini, and more and more examples are appearing in the literature, such as the binding of cofactors to photosynthetic membrane complexes [2], and the binding of a peptide to the OppA transport receptor [1].
Importantly, the new version of Martini, Martini 3, has opened the way to study protein-ligand binding on a much larger scale. Due to the improved description of small bead types, and the better representation of molecular geometries as well as cavities in proteins, the binding of small ligands to a variety of protein targets can now be faithfully (and very effciently !) captured with Martini, as shown in a recent proof of concept [3, see Figure]. A prospective of using this approach to build a high-throughput drug screening pipeline is presented in [4]. More examples of Martini 3 applied to predict protein-ligand interactions are found in [5].
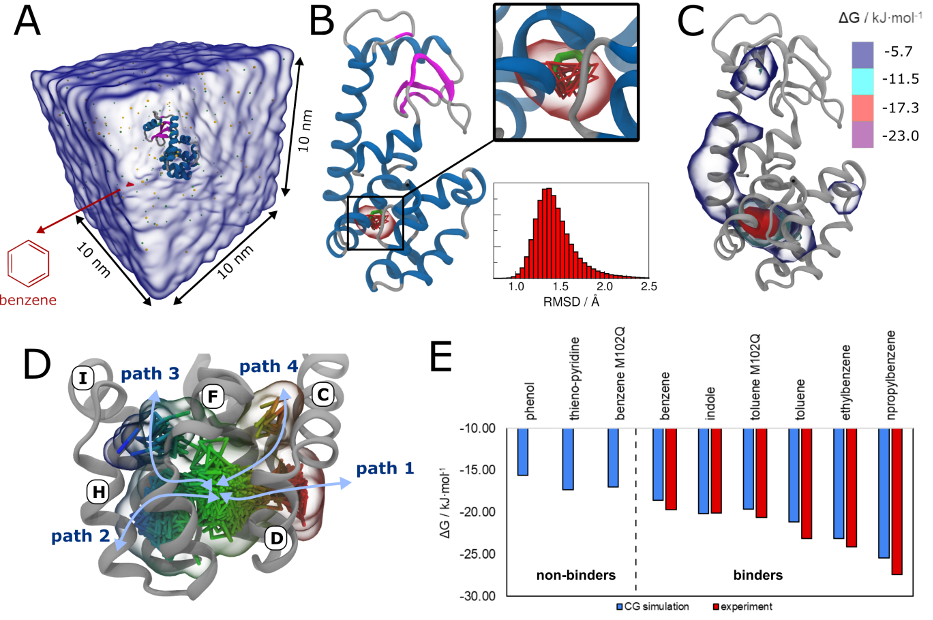
[1] R.P.A. Berntsson, M.K. Doeven, F. Fusetti, R.H. Duurkens, D. Sengupta, S.J. Marrink, A.M.W.H. Thunnissen, B. Poolman, D.J. Slotboom. The structural basis for peptide selection by the transport receptor OppA. EMBO J., 28:1332-1340, 2009. open access
[2] F.J. van Eerden, M.N. Melo, P.W.J.M. Frederix, X. Periole, S.J. Marrink. Exchange pathways of plastoquinone and plastoquinol in the photosystem II complex. Nature Commun. 8:15214, 2017. open access
[3] P.C.T. Souza, S. Thallmair, P. Conflitti, C. Ramírez-Palacios, R. Alessandri, S. Raniolo, V. Limongelli, S.J. Marrink. Protein–ligand binding with the coarse-grained Martini model. Nature Commun. 11:3714, 2020. doi:10.1038/s41467-020-17437-5
[4] P.C.T. Souza, V. Limongelli, S. Wu, S.J. Marrink, L. Monticelli. Perspectives on High-Throughput Ligand/Protein Docking With Martini MD Simulations. Front. Mol. Biosciences 8:657222, 2021. doi:10.3389/fmolb.2021.657222
[5] B. Waclawiková, P.C.T. Souza, M. Schwalbe, C.G. Neochoritis, W. Hoornenborg, S.A. Nelemans, S.J. Marrink, S. El Aidy. Potential binding modes of the gut bacterial metabolite, 5-hydroxyindole, to the intestinal L-type calcium channels and its impact on the microbiota in rats. Gut Microbes 15 (1), 2154544, 2023. doi:10.1080/19490976.2022.2154544



























































































































































 :
: